|
1,*
&
1,2Pedro Mendes
1Institute of Biological Sciences, Cledwyn Building, University of Wales, Aberystwyth SY23 3DD, UK
2
Present address:
National Center for Genome Resources,
1800-A Old Pecos Trail, Santa Fe, New Mexico 87505, U.S.A.
*Corresponding author: phone +44 161 2004492 fax +44 161 2004556,
email
http://dbk.ch.umist.ac.uk/
Summary |
Introduction |
Assumptions in MCA |
All cells are not the same |
Not all organisms are the same |
Moiety conservation and flux enhancement |
Modelling at the right scale |
Critique of top-down methods |
Proton-coupled electron transport-linked phosphorylation |
Large changes and MCA |
The not-very Universal Method |
Multi-site modulation |
Active learning |
Acknowledgements |
References
 |
Summary |
 |
|
|
|
|
We provide, as requested, a personal and critical survey
of some of the major limitations of the principles and applications of
metabolic control analysis, with special reference to the enhancement of fluxes
of biotechnological interest.
- Experimental methods of single-cell analysis such as flow cytometry
show that the implicit assumption that we study and model ensembles of
identical cells is completely untenable, and that cellular heterogeneity is
much greater than we normally assume.
- A feature of the post-genomic era is the recognition that many more
genes exist, and are expressed, than we had ever recognised, and that methods
are being developed for the quantitative asssessment of this. Even if the
individual flux-control coefficients of these gene products were each very
small their enormous number means that over-simplified analyses that ignore
them are very likely to lead to erroneous analyses of the true structure and
organisation of a metabolic system of interest.
- Even the assumption that the components of moiety-conserved cycles do
not change their total concentrations during an experiment ignores a
demonstrably large impact of this on the control structure. In the absence of
compartmentation or channelling, such cycles also serve to connect segments of
metabolism usually considered rather distant from each other.
- Simplified (‘top-down’) methods in which the system
structure is assumed a priori will not work for complex systems where
the combinatorial explosion of possible interactions requires much more
sophisticated methods for system identification.
- Dual-inhibitor titrations can reveal direct kinetic interactions
between individual catalytic activities in appropriate cases.
- No example exists in which one can extrapolate the conventional
control coefficients to provide reliable predictions about the behaviour of
metabolic systems subjected to ‘large’ changes in parameters.
- A "Universal Method" proposed for this (i) does not work when it is
the end product itself which feeds back to inhibit its own production
(as is usually the case in any high-yielding fermentation), and (ii) the method
works even in principle only for systems in which there are no
interactions between pathways, such as those involving conserved
moieties such as adenine and pyridine nucleotide couples.
Metabolic contol analysis and functional genomics share the same agenda,
in that they seek to relate the presence and activities of individual genes and
gene products to cellular biochemistry and physiology. They can be considered
to differ, however, in a philosophical sense since the former is essentially
deductive in character (and as practised) while the latter is of necessity
inductive, at least initially, because so many ORFs are of unknown function.
Inductive methods of machine learning should prove of value in unravelling
their properties.
|
|
Snapshots of Systems - Metabolic Control Analysis
and Biotechnology in the Post-Genomic Era
"So the first requirement will be for a theoretical framework in
which to embed all the detailed knowledge we have accumulated, to allow us to
compute outcomes of the complex interactions and to start to understand the
dynamics of the system. The second will be to make parallel measurements of the
behaviour of many components during the execution by the cell of an integrated
action in order to test whether the theory is right. Is there some other
approach? If I knew it I would be doing it, and not writing about the problem."
Sydney Brenner, 1997, in Loose Ends publ. Current Biology, London, p.73
|
|
|
 |
|
|
|
|
Holism and reductionism, and MCA’s view of the operations of complex biochemical systems
Following its original formulation in 1973
(Heinrich & Rapoport, 1973;
Heinrich & Rapoport, 1974;
Kacser & Burns, 1973)
as a means of understanding the
contribution of the individual steps of a biochemical pathway to the values of
flux and metabolite concentrations observed, some 13 years were to pass before
we first surveyed (Kell & Westerhoff, 1986a;
Kell & Westerhoff, 1986b) how the formalism, tools
and terms of metabolic control analysis might usefully be applied to such
systems in a biotechnological context. Since another such tridecennium has now
elapsed, it is timely to take stock of progress, to recognise that the take-up
of these methods among biotechnologists has been less than widespread, and (as
requested by the Editor) to give a personal and critical review
of successes, failures, problems and prospects for the use of MCA in
biotechnology. In what follows, it is taken that the reader has a good working
knowledge of the essential principles of MCA, as summarised for instance in
(Cornish-Bowden & Cárdenas, 1990;
Fell, 1992; Fell, 1996;
Heinrich & Schuster, 1996; Kell
et al., 1989; Kell & Westerhoff, 1986a;
Ovádi, 1995) and on the Internet at
http://dbk.ch.umist.ac.uk/mca_home.htm
and in links therefrom. In addition, we shall concentrate on unicellular
systems, implicitly those most commonly exploited to make products of
biotechnological interest.
Perhaps the chief intellectual benefits of MCA have been the recognition
(i) that in the steady state of a (linear) pathway where all steps are
proceeding at the same rate it is nevertheless appropriate to recognise that
each contributes quantitatively to the control of flux, in a manner which (for
small or infinitesimal changes) can be summed to unity, (ii) that the
flux-control coefficients so determined are consequently necessarily small, and
(iii) that the activities of many steps must be changed simultaneously if
fluxes are to be enhanced substantially. MCA thus constituted a bridge between
the rather reductionistic view then prevalent (that we can understand a systems
by looking at its component parts in isolation, without considering the
interactions between them - see (Kell & Welch,
1991; Mendes et al., 1995)) and the holistic
one (which in extremum - and in practice for many real, nonlinear,
coherent, self-organising systems (Kell & Hitchens,
1983) - would hold that the whole is so much more than the sum of its parts
that it is essentially pointless to consider the individual parts in isolation
at all (Ho, 1998)).
Coupled to these aperçus has been the recognition that computer
simulation can be a powerful tool in solving the forward problem of metabolism:
given the parameters of the system (usually the external metabolite and
effector concentrations and the kinetic properties of the enzymes) one can
solve the relevant differential equations and predict the time course and - if
such exist - the steady-state values of flux and metabolite concentrations.
Software such as the program gepasi
produced in aberystwyth by one of us (Mendes, 1993;
Mendes, 1997; Mendes & Kell,
1998a, b) has been designed for (and indeed by)
biologists (and successfully hides the mathematical details from the typical
user), and given a simulation of a pathway it is easy to extract the ‘MCA
properties’ such as flux- and concentration-control coefficients by
numerical simulation of the effects of small changes in parameter values or
analytically by differentiating the rate equations to acquire the elasticities
and inverting the elasticity matrix so obtained
(Fell, 1996;
Fell & Sauro, 1985;
Mendes, 1993;
Reder, 1988;
Westerhoff & Kell, 1987).
The metabolic control anaysis of a system is thus normally ‘merely’ a
snapshot of a typically rather restricted subset of the cellular biochemistry
actually taking place in time and space.
|
|
|
|
 |
|
|
|
|
Assumptions in MCA, implicit and explicit
With its concentration on small or infinitesimal changes, a domain where
for (spatially) homogeneous systems its analysis is both exact and complete,
MCA necessarily represents an approximation to a more complex reality,
and this begs the question of how adequate this approximation is. In view of
the recognition that it is but a subset of a full simulation of whatever system
it is desired to simulate, it is probably unsupportable. Some other common
assumptions of MCA (and its usual implementations) which will be explored later
are summarised in Table 1. They include the implicit view that all cells in a
suspension are the same, that it is possible to lump together large segments of
metabolism without losing important knowledge of the behaviour of the overall
system, and that there are ‘universal’ methods which can permit the
rational optimisation of metabolic fluxes in systems of arbitrarily complex
organisation.
Table 1. Some explicit and
implicit assumptions of modern MCA, and some inadequacies of its usual
implementations in a biotechnological context.
| Assumption / Misapplication |
Comments / Consequences |
| All cells are the same |
Heterogeneity is very much greater than normally assumed, and
this can be determined experimentally using single-cell analyses
|
| Simple models are adequate |
Genome sequencing has uncovered the fact that we know the function of
fewer than half of their genes, and there is evidence that almost all
contribute to fitness even in laboratory conditions. Much more of
metabolism is relevant to a flux than is normally recognised.
|
|
The Universal Method permits a rational approach to
the optimisation of flux in any metabolic system
|
It doesn’t work if (i) the end-product feeds back to inhibit its
own synthesis, whetehr kinetically or by mass action, and/or (ii) there
are interactions between pathway branches involving moiety-conserved
cycles.
|
|
The coefficients of MCA determined using large changes are not too badly
different from those obtained via very small parameter changes
|
Nonlinearities, unknown interactions and the overall complexity of
biological systems mean that deviation indices are reasonably
small only in simple systems.
|
|
|
|
|
|
|
|
|
|
All cells in an axenic culture are not the same; microbial differentiation
Whilst it is rather obvious that the phenotypes of all cells in a
differentiated organism are not the same (so much so that
there is no such thing as a biochemically "normal"
individual (Williams, 1956)), it is
implicit in a standard MCA analysis that they are; in other words
we tend to treat the system under study as an ensemble in the
thermodynamic sense (Welch & Kell, 1986;
Westerhoff & van Dam, 1987). In
fact, the essence of the problem (Kell et al.,
1991) is that one is trying, typically, to correlate a rate of change (v)
of a certain variable with respect to the value of a certain parameter or
property (p), but a correlation may be expected between the mean values and
only if v is kinetically of first order with respect to p. This
is completely unrealistic even for the axenic microbial cultures that are the
focus of this review, and with the availability of techniques such as flow
cytometry (Davey & Kell, 1996;
Kell et al., 1991) it becomes possible to determine
the heterogeneity of cellular properties directly. In one example of our
own (Kaprelyants & Kell, 1992), the extent to
which chemostat-grown (and thus as near as one can get to genuinely
steady-state cultures of) Micrococcus luteus cells could take up the
membrane energisation probe rhodamine 123 varied by more than 1000-fold under
conditions in which uptake was fully uncoupler-sensitive and neither efflux
pumps nor lack of membrane permeability were an issue. The unwanted consequence
of the failure to take culture heterogeneity into account (in terms of being
led to erroneous conclusions about causality and mechanism) reaches its
apotheosis in the study of microbial viability/culturability
(Kell et al., 1998). Such an analysis ignoring
heterogeneity will also tend to mask intercellular interactions
(Fuqua et al., 1996;
Kell et al., 1995)
such as those in which culturable cells secrete a factor necessary
for the resuscitation and growth of non-growing cells of the same organism
(Kaprelyants & Kell, 1993;
Kaprelyants et al., 1994;
Kaprelyants et al., 1999;
Mukamolova et al., 1998).
|
|
|
|
|
|
|
|
|
Not all organisms are the same...
So far as the typical textbook of biochemistry is concerned, cells are
inevitably taken to be essentially similar, with a great majority of their
broad activities (and the ‘housekeeping’ genes which code for them)
being common throughout biology (at least at the level of prokaryote, eukaryote
and archaean). Specific features such as photsynthesis or nitrogen fixation are
seen merely as occasional adjuncts. Our attempts to simulate metabolism seem to
rely implicitly on this, and it is perhaps assumed that models have a validity
beyond the sytem for which they are constructed. However, as we enter the
post-genomic era, two major facts have become evident; (i) many or most ORFs
code for products of unknown function
(Blattner et al., 1997;
Bork et al., 1998;
Cole et al., 1998;
Goffeau et al., 1996;
Hinton, 1997;
Oliver, 1996)
with many being conserved but most comparatively unique
(Koonin & Galperin, 1997),
and (ii) large-scale, genome-wide comparisons of orthologous genes point up the
prevalence of horizontal gene transfer
(Forterre, 1997a;
Forterre, 1997b;
Koonin et al., 1997;
Rivera et al., 1998)
and the consequent inadequacy of gradualist views of evolution.
(Note however that these analyses are to date restricted to the very
small fraction (Amann et al., 1995) of cultured
microbes, and that many close relative of existing taxa remain to be cultured
(Kaprelyants et al., 1999;
Kell et al., 1998;
McVeigh et al., 1996).)
Consequently, we now recognise that many more genes contribute to fitness
than had previously been considered.
The major approaches to functional genomics currently being undertaken
involve the systematic knocking out of individual genes seriatim;
where this is being done, e.g. in S. cerevisiae
(Dujon, 1998;
Oliver & Baganz, 1998;
Oliver et al., 1998;
Teusink et al., 1998),
it is found that only some 15% are ‘essential’, and the
question arises as to the role of the others - do they have a very
high contribution to fitness under a restricted set of conditions
met only occasionally and never in the laboratory, or do they
all provide a marginal contribution to fitness? At least as judged by the fact
that they are both transcribed under laboratory conditions in rich media and
can be shown to contribute to fitness in sensitive growth rate tests
(Eisen et al., 1998;
Smith et al., 1995;
Smith et al., 1996;
Thatcher et al., 1998),
one is led to attach
significance to the latter view. The mental picture which emerges then is that
whilst there may be core or major blocks of primary metabolism which are
important, the contribution of the rest of the cellular activities which are
normally neglected is likely to be just as great or greater. Although they may
be individually small they are collectively numerous (the totals of genes in
E. coli, baker’s yeast and Streptomyces coelicolor
A3(2) are some 4000, 6000 and 8000 respectively), and while fewer will
contribute to a flux than to the overall fitness as correctly judged
(Kell, 1987;
Westerhoff et al., 1983)
by growth rate, the emerging paradigm is of a much
greater complexity and sophistication of unicellular controls than we had
heretofore recognised. One example of an important (and probably excessive)
simplification in common usage is that the total values of pyridine nucleotides
in cellular compartments are not of particular significance.
|
|
|
|
|
|
|
|
|
Moiety conservation and flux enhancement
One aspect of metabolism that has received little attention for the
purposes of flux maximisation is the existence of moiety-conserved cycles
(Reich & Sel'kov, 1981). These are ubiquitous in
metabolism and a few of them (e.g. NAD/NADH and ATP/ADP) act as major links
between various parts of metabolism. Whilst affecting the poise of these
cofactor couples can be most useful in metabolic engineering
(Lopez de Felipe et al., 1998)
the flux through a pathway, as seen within the MCA formalism
(Hofmeyr et al., 1986), is also controlled by
the total amount of conserved moieties. We have observed by computer
simulation that the flux of several model pathways responds to the total amount
of conserved moiety according to a bell-shaped curve. This suggests that there
is an optimal amount of cofactor for a given pathway flux (and that
compartmenation of pathways would be necessary to optimise them separately),
otherwise the flux will be somewhat limited by the availability of the
conserved moiety. To manipulate the total amount of the moiety we will thus
need to target the pathways of their biosynthesis and degradation.
Alternatively we could as well manipulate the number and/or affinity of moiety
binding sites which modulate the amount of available (i.e. free) total
moiety. The existence of large numbers of NAD binding sites may indeed be a
cellular mechanism for the rapid adjustments of the available total amount of
NAD (rather than a slower adjustment by increased/decreased biosynthesis). Fig.
1 depicts such a bell-shaped relation between the total amount of the moiety
and the flux for a simple branched pathway in which other parameters are held
constant.
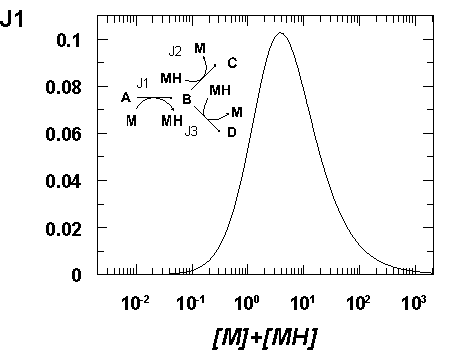 |
|
Figure 1. Dependence of entry steady-state flux on the total amount
of conserved moiety in a model branched pathway. The pathway simulated is
shown in the inset, the arrows representing the positive direction of flux.
All reactions are fully reversible, all kinetic and equilibrium constants
are unity, [A] = 10, [C] = [D] = 1 such that
the overall disequilibrium ratio is 0.1 on both branches.
|
|
|
|
|
|
|
|
|
|
On modelling at the right scale
We have traditionally treated our systems relatively simply, due in part
to the difficulty of measuring everything. With the emergence of measurements
of the proteome (Anderson & Anderson, 1998;
Wilkins et al., 1997), the transcriptome
(de Saizieu et al., 1998;
DeRisi et al., 1997;
schena et al., 1996;
Spellman et al., 1998;
Velculescu et al., 1997;
Wodicka et al., 1997)
and the metabolome
(Oliver & Baganz, 1998;
Oliver et al., 1998)
we now have the ability to carry out hundreds of measurements on
macromolecular and metabolic variables simultaneously.
The outcomes of the pioneering studies are in many cases given in the form of
lists of expression ratios for the hundreds of genes of interest, which are
hard to interpret - the appropriate scale for easy understanding is not a
life-sized model (Eisen et al., 1998). Treating
related segments of metabolism as ‘blocks’ is one solution
(Kell et al., 1989),
sometimes referred to as top-down analysis
(Brand, 1996;
Brand, 1998;
Brown et al., 1990)
and is being exploited in functional genomics as the FANCY method
(Oliver & Baganz, 1998;
Teusink et al., 1998),
but this approach fails to give a true account of the system of interest
under a number of circumstances.
|
|
|
|
|
|
|
|
|
A critique of ‘top-down’ methods in which segments of metabolism are treated as one
In the early literature of MCA (Kacser & Burns,
1973) it was already proposed that one could group sequential enzymes and
treat them as one unit for the purposes of control analysis. This is possible
due to the summation theorem (Kacser & Burns, 1973)
and the fact that the elasticity concept can be applied to groups of reactions
(Kacser, 1983). Brown et al. (Brown et al., 1990) took this one step further and
formally proposed the ‘top-down’ method for determining control
coefficients. In this method one builds two groups of metabolic steps around
(upstream and downstream of) one single intermediate metabolite. Provided that
this central intermediate metabolite is the only kinetic link between the two
groups of steps then one can determine the elasticites of the two groups
towards that metabolite with just two single-modulation experiments. Group
control coefficients can then be calculated using the connectivity and
summation theorems (the assumption therefore being that no other interactions -
such as feedback loops or metabolic channelling (Agius &
Sherratt, 1997; Mendes et al., 1995;
Ovádi, 1995) - exist between the two groups). The
control coefficients thus obtained for the two groups of steps could reveal
which of them has more control, e.g. as in (Simpson et al.
1998; Stephanopoulos & Simpson, 1997),
where knowledge of this can allow one to vary the control structure of a
pathway to improve fluxes of biotechnological interest (Simpson et al., 1995).
An attractive application of this method would be to apply it repeatedly
by subdividing each group into smaller groups. This would result in the
determination of the complete control structure of the pathway, each time with
more resolution (hence the ‘top-down’ name). Although elegant in
conception, this method is effectively impossible in practice, due to the
requirement of grouping steps such that there is only one kinetic link (via the
intermediate metabolite) between them. Ainscow and Brand have recently extended
the method (Ainscow & Brand, 1995) such that it
can be applied to the case when the groups of steps are connected by more than
one metabolite. Nevertheless, for the method to work as intended one must be
absolutely sure that all the kinetic links between the groups of steps are
known and included in the analysis explicitly (or are unaffected by the
modulations performed (Ainscow & Brand, 1998a)).
Thus, one can never be sure that the coefficients determined by this method are
correct as there could be extra kinetic interactions between the two groups of
steps other than the ones taken into consideration. The classical method of
direct determination of control coefficients by perturbation of enzyme
activities is immune from this problem and so could (and should) be used to
confirm the results with the top-down method - but this rather defeats the
purpose of using it in the first place! (Recently, Kholodenko and colleagues
have presented a combination of top-down MCA and the perturbation method which
they refer to as ‘Metabolic Design Analysis’ (Kholodenko et al., 1998).) But there are extra
problems when one wants to use this method in general: (i) there are several
known (and certainly many more unknown) feedback loops in metabolism and
(ii) many reactions include cosubstrates such as NAD/NADH or ATP/ADP which form
kinetic links between steps normally considered distant. Both these reduce the
number of metabolites that can be effectively used in the top-down approach to
separate groups of steps. As such the method is not amenable, in general, to a
true ‘top-down’ approach of measuring all control
coefficients. The problems are greatly compounded by the propagation of errors
and bias contingent on the measurement of elasticities (Schlosser et al., 1993; Thomas
& Fell, 1995) and control coefficients (Ehlde &
Zacchi, 1996; Small, 1993), and such errors may not
be normally distributed (Ainscow & Brand, 1998b).
We therefore find that the (correct) application of the top-down method to
large, complex biosystems may be rather limited in practice, and above all
dangerous if results are not validated by an independent method.
Note that this is not a critique of simplification per se, since
in many cases the intrinsic dimensonality of the major blocks of a complex
system of interest may well be comparatively small and the level of
understanding that we require, and indeed good precision in our models (Broadhurst
et al., 1997; Kell & Sonnleitner, 1995;
Shaw et al., 1997), is more easily attained with
small models than with large ones. But this is something that we find out
afterwards, when measurements of many variables have been made
and evaluated (Eisen et al., 1998), not something
to build in beforehand!
|
|
|
|
|
|
|
|
|
Proton-coupled electron transport-linked phosphorylation - an example
of a chanelled system, assessed using dual-inhibitor titrations
A consequence of lumping reactions together in the macroscopic way
typified by the top-down approach is that it is assumed that their
intermediates are delocalised. One of the major areas of interest of this
laboratory has been in the problem of channelling, most recently in terms of
intermediary metabolites (Mendes et al.,1992 ,
1995, 1996) but more
classically in terms of the problem of whether the energetic intermediates of
electron transport-linked phosphorylation are delocalised or not
(Kell, 1979;
Kell, 1988;
Kell & Westerhoff, 1990).
The basic idea is as follows. In the classical chemiosmotic coupling model
(Mitchell, 1966;
Nicholls & Ferguson, 1992),
electron transport generates a transmembrane proton
gradient which, due to the rapid diffusion rates of protons in aqueous media,
leads to a delocalised protonmotive force Dp
consisting of a membrane potential Dy and a pH
gradient zDpH which is consequently avaiable to
all ATP synthase enzymes in the organelle in whose membrane the
pmf-generators are embedded. Uncouplers act by dissipating the pmf as heat. (In
addition, the pmf can in principle feed back to inhibit electron transport via
‘slip’ or be dissipated ‘naturally’ to heat via a
pmf-dependent ‘leak slip’ which does not differ formally from the
imperfect coupling naturally present.) This is depicted in Fig 2.
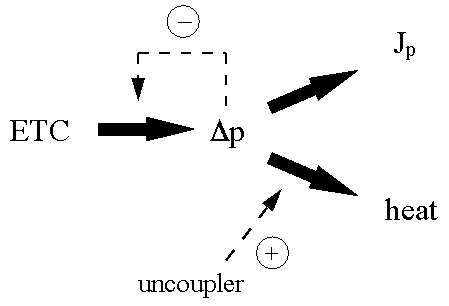 |
|
Figure 2. The classical chemiosmotic coupling paradigm for
electron transport-linked phosphorylation.
|
The consequence is that the rate of phosphylation Jp depends only and
monotonically on Dp, typically according (when
assessed experimentally by acid-bath-type experiments) to a sigmoidal function
of the pmf, i.e.:
|
Jp = [ATP-synthase] . f(Dp)
|
(Eq. 1) |
In a typical dual-inhibitor titration using an uncoupler and an ATP
synthase inhibitor, we first study the effect of uncoupler on Jp, with results
similar to those in Fig 3 (open circles). We then inhibit half of the ATP
synthases using a tight-binding (or better covalent) inhibitor such that Jp
falls to one half of its original value (Fig 3). The effect of the uncoupler
titration that must be predicted from the delocalised chemiosmotic type of
uncoupling model is similar to that given by the closed triangles in Fig 3,
since the pmf canot be made smaller by this treatment and it is probably
slightly larger due to the smaller drain on it - the shape of the curve is the
same but the rate at any level of uncoupler (and putatively pmf) is just one
half of the control. Similarly, the amount of uncoupler needed to achieve full
uncoupling is the same. Unfortunately for this view, the experimental curve is
quite different: lowering the initial Jp to one-half of its original value with
the ATP synthase inhibitor decreases by one half the amunt of uncoupler
necessary to achieve full uncoupling. No delocalised coupling
model can account for this type of behaviour in uncoupler/energy transfer
inhibitor titrations
(Herweijer et al.,1986;
Hitchens & Kell, 1983a;
Hitchens & Kell, 1983b;
Kell, 1988;
Kell, 1992;
Westerhoff & Kell, 1988),
and indeed none has made a serious attempt to do so.
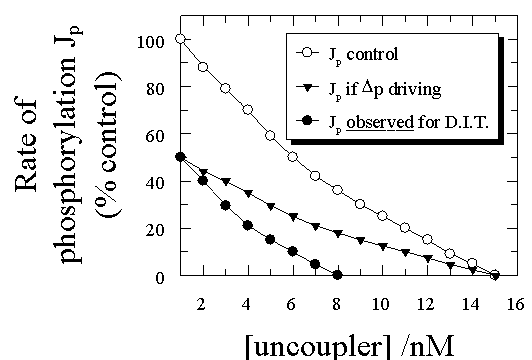 |
|
Figure 3. Theoretical and experimental traces from dual
uncoupler/energy transfer inhibitor titrations.
|
|
|
|
|
|
|
|
|
|
Between the Bud and the Rose: Large Changes and Metabolic Control Analysis
As is well-known, and is mentioned above, the theorems of classical MCA
work only for parameter changes which are small (and in principle
infintesimal). However, it is to be assumed (and see below) that substantial
increases in flux towards metabolites of biotechnological interest require
large changes in at least some of the parameters such as enzyme concentrations
(and the same is true for the phenotypic manifestation of disease states when a
sufficient (‘theshold’) loss in enzymatic function is induced
(Durrieu et al., 1997;
Letellier et al., 1998;
Mazat et al., 1997,
1998)).
Small and Kacser tackled the problem of exactly how great an inaccuracy in
estimating control coefficients via large changes in parameters might be
involved. They first introduced the idea of a deviation index as
the relative change in a metabolic variable such as a flux to a large
change in a parameter (Small & Kacser, 1993a),
and showed that for unbranched chains of enzymes with linear kinetics there
was a direct relationship between deviation indices and flux-control
coefficients. They also pointed out that combined changes of the activity
of individual enzymes will produce a more-than-additive response (and see
below). The behaviour of branched and non-linear pathways was more complex
(Small & Kacser, 1993b), and though it was stated
that many metabolic systems behave in practice as quasi-linear systems, the
differences between the actual and predicted amplification factors were often
quite great. Indeed, a detailed study by R. Schuster & Holzhütter
(Schuster & Holzhutter, 1995) of erythrocyte
properties resulting from large-scale alterations in enzymatic activities
concluded that no existing extrapolation method using the conventional
control coefficients was able to provide reliable predictions.
|
|
|
|
|
|
|
|
|
The not-very Universal Method
It is worth noting that, as proven in the summation theorem of Metabolic
Control Analysis (e.g.
Cornish-Bowden et al., 1995;
Fell, 1996;
Heinrich & Rapoport, 1974;
Heinrich & Schuster, 1996;
Kacser & Burns, 1973;
Kell & Westerhoff, 1986a),
changes in the concentrations of individual enzymes tend to have little
effect on particular metabolic fluxes (nor, indeed, on the gross phenotype
under most laboratory conditions
(Thatcher et al., 1998)).
However, in part because of the so-called connectivities of MCA, changes
in individual enzyme concentrations can and do have substantial effects
on metabolite concentrations, even when the changes in flux are
negligible
(Mendes et al., 1995;
Mendes et al., 1996).
It is therefore very reasonable that attempts to increase metabolic fluxes
by increasing the concentrations of metabolic enzymes
may lead to substantial increases in metabolite levels, and that these may
either prove cytotoxic or at least necessarily lead to the diversion of flux to
pathways other than that desired. It would therefore be desirable (if it were
indeed possible) to seek to modulate fluxes by changing enzyme activities in a
manner that managed to preserve the steady-state levels of metabolites.
Thus Kacser and Acerenza (Kacser & Acerenza, 1993)
introduced the so-called Universal Method that purported to have this effect
and to be ‘entirely general’. In the Universal Method, it is
recognised that in any pathway leading to the output of interest, the
activities of whose enzymes one would wish to increase, there will be branch
points leading to other parts of metabolism which should not be perturbed.
Because of the conservation of mass, the fluxes down each branch point
following a change in flux are related both to the changes in flux before and
after the branchpoint in the ‘main’ pathway and to the ratio r of
enzyme activities before and after the change in flux. For unimolecular
reactions, there is a unique value of r for each such reaction at which the
fluxes down the branches remain unchanged.
However, this method has two major failings: (i) it does not work when
it is the end product itself which feeds back to inhibit its own
production (and this is usually the case in any high-yielding fermentation, for
both thermodynamic and kinetic reasons), and (ii) the method works in principle
only for systems in which there are no interactions between pathways,
such as those involving conserved moieties such as adenine and pyridine
nucleotide couples. Since any system of interest is likely intimately to
involve cofactors of this type, it seems that the Universal Method as presented
is unlikely to prove of substantive utility, nor seems to have done so to date.
|
|
|
|
|
|
|
|
|
Multi-site modulation
Whilst the Universal Method cannnot work as advertised, it does draw
attention to the need - whatever the effects on the rest of metabolism -
for multisite modulations to be performed if there is to be a substantial
increase in flux, and this is now widely recognised
(Cornish-Bowden, 1995;
Cornish-Bowden et al., 1995;
Fell, 1998;
Fell & Thomas, 1995;
Niederberger et al., 1992;
Thomas & Fell, 1998).
This does not contradict any of the insights of MCA, and in fact it can
be shown both by analysis and simulation
(Small & Kacser, 1993a,
1993b)
that this result is to be expected: as soon as the step with higher control
becomes faster (as happens with overexpression) the control shifts to other
steps in the pathway. It is now evident that for any strategy to be successful
in increasing the flux of a pathway substantially there is a requirement for
the manipulation of several steps. This was clearly demonstrated
experimentally by Niederberger and colleagues in their classical study
(Niederberger et al., 1992) and has been
discussed at some length by Fell and Thomas
(Fell, 1998;
Fell & Thomas, 1995;
Thomas & Fell, 1998).
In a recent conference it was evident that the metabolic engineering community
(both research and industry) is converging to this conclusion, and it is now
largely accepted that to increase flux one should manipulate at least two
metabolic steps (Mendes & Kell, 1997). Removing
the fluxes to unproductive pathways is likely to be much more significant for
mature fermentations than seeking solely to stimulate the flux through the
desired one per se
(Holms, 1996;
Holms et al., 1991).
|
|
|
|
|
|
|
|
|
Active learning and a post-Baconian approach to science in the post-genomic era
The commonest conventional method of experimental science, generally
referred to as ‘the scientific method’, involves the preparation of
an experimental system in a specified state and the manipulation of,
preferably, a single parameter, whereupon one observes the time-evolution of
the values of one or more variables compared to that of a control in which the
‘triggering’ manipulation is not performed. The parameter may then be
moved to different set points. Each of those variables might also be controlled
at a fixed level, i.e. as a parameter, and comparable experiments performed. If
the system is comparatively simple and well behaved (e.g. asymptotically
stable, and not chaotic as in (Davey et al., 1996))
it is usually possible to determine the form and parameterisation of the
equations relating the parameters and the variables and the time evolution
of the system by mathematical fitting procedures (Mendes
& Kell, 1998a), leading to what is usually considered an
"understanding" of the system. However, this is true only for
simple systems, and one may put forward the views that (i) this kind of
deductive reasoning is that usually practised in the MCA community, (ii)
the functional genomics agenda
(Bork et al., 1998;
Hieter & Boguski, 1997;
Hinton, 1997;
Kell, 1998;
Oliver, 1996)
is likely to be much better attacked under current conditions via an
inductive type of approach.
Indeed, we consider that complex systems cannot be treated to best advantage
(Westerhoff & Kell, 1996)
in this more classical, deductive way. First of all, there are far too many
variables and potential parameters for an exhaustive set of experiments to
be performed, and those parameter sets producing ‘desirable’
outcomes may be few and far between. (For n parameters which might
adopt m values the number of combinations is obviously
mn, and even if m is only a miserable 2 for
n=100, 2100 ~ 1031, and the lifetime of the
Universe is ‘only’ some 1017s.) The inevitable
conclusion for the study of complex systems is that we we must vary many
(or at least several) parameters at a time and use the methods of multivariate
statistics and machine learning to deconvolute the data so obtained to extract
those features most relevant to the operation of the system. Then, because of
the high dimensionality of the system and problem, we must iterate this process
further (somewhat in the way in which we traditionally need to provide
rounds of mutation and selection in fermentation development programmes
(Crueger & Crueger, 1989)).
Indeed, our own approach in recent years to the understanding of complex
cellular systems has been to exploit spectroscopic methods such as pyrolysis
mass spectrometry
(Broadhurst et al., 1997;
Gilbert et al., 1997;
Goodacre & Kell, 1996;
Goodacre et al., 1993,
1994a,
1994b,
1996a;
Taylor et al., 1998),
ft-ir
(Goodacre et al., 1996b,
1998a;
Oliver et al., 1998;
Winson et al., 1997,
1998),
and dispersive Raman
(Goodacre et al., 1998b)
in which hundreds of variables are measured simultaneously, and to
couple these measurements with advanced chemometric and related analyses
based on the methods of artificial intelligence, machine learning and
evolutionary computing
(Bäck et al., 1997;
Michie et al., 1994;
Rich & Knight, 1991;
Weiss & Kulikowski, 1991).
Although the above described the overall structure of a single
experiment, scientific research of course proceeds by a process
of experimental hypothesis testing (e.g. Oldroyd, 1986),
and it is appropriate to end by outlining one way of computer-assisted
knowledge acquisition with which we think important progress might be made.
This process is an active approach, which in fact differs markedly from the
passive nature of most ‘scientific discovery’ systems
(Langley et al., 1987),
which either receive data all at once in a single batch, or have no choice
over the next example (Raju & Cooney, 1998),
and suffer from the problem that most of the observables have little bearing
on the overall outcome (Blum & Langley, 1997)
and for the purposes of this analysis amount to ‘noise’. The study
of systems that can choose the next experiment is known as
‘active learning’. There are two computational tasks in
active learning: formation of hypotheses that are consistent with known
background knowledge and experimental results, and selection of the best
experiment (or set of experiments) to discriminate between hypotheses. It
should be noted that experiment selection in active learning is not to be
confused with the traditional statistical study of experimental design, where
the difference is between deciding which question to ask next (active learning)
versus ensuring that a set of experiments can answer a question (traditional
experimental design).
To conclude, it seems reasonable that active learning approaches can
lead us efficiently to means for asking and answering the right kinds of
question at the right kind of complexity in the post-genomic era.
|
|
|
Publications |
|
|
|
|
|
Acknowledgments
We thank Ross King and Steve Oliver for useful discussions, and the BBSRC for financial support.
|
|
|
Publications |
|
|
|
|
|
References
Agius, L. & Sherratt, H. S. A. (1997). Channelling in intermediary metabolism. Portland Press, London. [synopsis]
Ainscow, E. K. & Brand, M. D. (1995).
Top-down control analysis of systems with more than one common intermediate.
Eur. J. Biochem. 231, 579-586. [
abstract
]
Ainscow, E. K. & Brand, M. D. (1998a).
Control analysis of systems with reaction blocks that 'cross-talk'. Biochim.
Biophys. Acta 1366, 284-290. [
abstract
]
Ainscow, E. K. & Brand, M. D. (1998b). Errors associated with metabolic control analysis. Application of Monte Carlosimulation of experimental data. J. Theoret. Biol. 194, 223-233. [abstract]
Amann, R. I., Ludwig, W. & Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143-169. [abstract]
Anderson, N. L. & Anderson, N. G. (1998). Proteome and proteomics: New technologies, new concepts, and new words. Electrophoresis 19, 1853-1861. [abstract]
Bäck, T., Fogel, D. B. & Michalewicz, Z. (1997). Handbook of evolutionary computation. IOPPublishing/Oxford University Press, Oxford. [synopsis]
Blattner, F. R., Plunkett, G., Bloch, C. A., Perna, N. T., Burland, V., Riley, M., ColladoVides, J., Glasner, J. D., Rode, C. K., Mayhew, G. F., Gregor, J., Davis, N. W., Kirkpatrick, H. A., Goeden, M. A., Rose, D. J., Mau, B. & Shao, Y. (1997). The complete genome sequence of Escherichia coli K-12. Science 277, 14531462. [abstract]
Blum, A. L. & Langley, P. (1997). Selection of relevant features and examples in machine learning. Artificial Intelligence 97, 245-271.
Bork, P., Dandekar, T., Diaz-Lazcoz, Y., Eisenhaber, F., Huynen, M. & Yuan, Y. P. (1998). Predicting function: From genes to genomes and back. J. Mol. Biol. 283, 707-725. [abstract]
Brand, M. D. (1996). Top down metabolic control analysis. J. Theoret. Biol. 182, 351-360. [abstract]
Brand, M. D. (1998). Top-down elasticity analysis and its application to energy metabolism in isolated mitochondria and intact cells. Mol. Cell. Biochem. 184, 13-20. [abstract]
Broadhurst, D., Goodacre, R., Jones, A., Rowland, J. J. & Kell, D. B. (1997). Genetic algorithms as a method for variable selection in multiple linear regression and partial least squares regression, with applications to pyrolysis mass spectrometry. Anal. Chim. Acta 348, 71-86.
Brown, G. C., Hafner, R. P. & Brand, M. D. (1990). A top-down approach to the determination of control coefficients in metabolic control theory. Eur. J. Biochem. 188, 321-325.
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S., Barry, C. E., Tekaia, F., Badcock, K., Basham, D., Brown, D., Chillingworth, T., Connor, R.,
Davies, R., Devlin, K., Feltwell, T., Gentles, S., Hamlin, N., Holroyd, S., Hornby, T., Jagels, K., Krogh, A., McLean, J., Moule, S., Murphy, L., Oliver, K., Osborne, J., Quail, M. A., Rajandream, M. A., Rogers, J., Rutter, S., Seeger, K., Skelton, J., Squares, R., Squares, S., Sulston, J. E., Taylor, K., Whitehead, S. & Barrell, B. G. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537-544.
[abstract]
Cornish-Bowden, A. (1995). Kinetics of multi-enzyme systems. In Biotechnology, vol. 9 (ed. H. J. Rehm, G. Reed, A. Pühler and P. Stadler), pp. 121-136. Verlag Chemie, Weinheim.
Cornish-Bowden, A. & Cárdenas, M. L. (1990). Control of metabolic processes. Plenum Press, New York. [contents]
Cornish-Bowden, A., Hofmeyr, J.-H. S. & Cárdenas, M. L. (1995). Strategies for manipulating metabolic fluxes in biotechnology. Bioorg. Chem. 23, 439-449. [abstract]
Crueger, W. & Crueger, A. (1989). Biotechnology: a textbook of industrial microbiology. Sinauer Associates, Sunderland, MA.
Davey, H. M., Davey, C. L., Woodward, A. M., Edmonds, A. N., Lee, A. W. & Kell, D. B. (1996). Oscillatory, stochastic and chaotic growth rate fluctuations in permittistatically-controlled yeast cultures. Biosystems 39, 43-61. [abstract]
Davey, H. M. & Kell, D. B. (1996). Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analysis. Microbiol. Rev. 60, 641-696. [abstract]
de Saizieu, A., Certa, U., Warrington, J., Gray, C., Keck, W. & Mous, J. (1998). Bacterial transcript imaging by hybridization of total RNA to oligonucleotide arrays. Nature Biotechnol. 16, 45-48. [abstract]
DeRisi, J. L., Iyer, V. R. & Brown, P. O. (1997). Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680-686. [abstract]
Dujon, B. (1998). European Functional Analysis Network (EUROFAN) and the functional analysis of the Saccharomyces cerevisiae genome. Electrophoresis 19, 617-624. [abstract]
Durrieu, G., Letellier, T., Antoch, J., Deshouillers, J. M., Malgat, M. & Mazat, J. P. (1997). Identification of mitochondrial deficiency using principal component analysis. Mol. Cell. Biochem. 174, 149-156. [abstract]
Ehlde, M. & Zacchi, G. (1996). Influence of experimental errors on the determination of flux control coefficients from transient metabolic concentrations. Biochem. J. 313, 721-727. [abstract]
Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95, 14863-14868. [abstract]
Fell, D. A. (1992). Metabolic Control Analysis - a survey of its theoretical and experimental development. Biochem. J. 286, 313-330.
Fell, D. A. (1996). Understanding the Control of Metabolism. Portland Press, London. [
synopsis]
Fell, D. A. (1998). Increasing the flux in metabolic pathways: A metabolic control analysis perspective. Biotechnol. Bioeng. 58, 121-124.
Fell, D. A. & Sauro, H. M. (1985). Metabolic control and its analysis. Additional relationships between elasticities and control coefficients. Eur. J. Biochem. 148, 555-561. [abstract]
Fell, D. A. & Thomas, S. (1995). Physiological control of metabolic flux: the requirement for multisite modulation. Biochem. J. 311, 35-39. [abstract]
Forterre, P. (1997a). Archaea: what can we learn from their sequences? Curr. Op. Genet. & Develop. 7, 764-770. [abstract]
Forterre, P. (1997b). Protein versus rRNA: Problems in rooting the universal tree of life. ASM News 63, 89-95.
Fuqua, C., Winans S.C. & Greenberg, E.P. (1996). Census and consensus in bacterial ecosystems: The LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu. Rev. Microbiol. 50, 727-751. [
abstract] [full text]
Gilbert, R. J., Goodacre, R., Woodward, A. M. & Kell, D. B. (1997). Genetic programming: A novel method for the quantitative analysis of pyrolysis mass spectral data. Anal. Chem. 69, 4381-4389. [abstract (requires subscription)]
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J. D., Jacq, C., Johnston, M., Louis, E. J., Mewes, H. W., Murakami, Y., Philippsen, P., Tettelin, H. & Oliver, S. G. (1996). Life With 6000 Genes. Science 274, 546-567. [abstract]
Goodacre, R. & Kell, D. B. (1996). Pyrolysis mass spectrometry and its applications in biotechnology. Curr. Opinion Biotechnol. 7, 20-28. [abstract] [full text]
Goodacre, R., Kell, D. B. & Bianchi, G. (1993). Rapid assessment of the adulteration of virgin olive oils by other seed oils using pyrolysis mass spectrometry and artificial neural networks. J. Sci. Food Agri. 63, 297-307. [abstract] [condensed paper]
Goodacre, R., Neal, M. J. & Kell, D. B. (1994a). Rapid and quantitative analysis of the pyrolysis mass spectra of complex binary and tertiary mixtures using multivariate calibration and artificial neural networks. Anal. Chem. 66, 1070-1085. [abstract]
Goodacre, R., Neal, M. J. & Kell, D. B. (1996a). Quantitative analysis of multivariate data using artificial neural networks: a tutorial review and applications to the deconvolution of pyrolysis mass spectra. Zentralblatt. für Bakteriologie 284, 516-539. [abstract] [full paper]
Goodacre, R., Rooney, P. J. & Kell, D. B. (1998a). Rapid analysis of microbial systems using vibrational spectroscopy and supervised learning methods: application to the discrimination between methicillin-resistant and methicillin-susceptible Staphylococcus aureus. In Proc. SPIE (Infrared Spectroscopy: New Tool in Medicine), vol. 3257 (ed. M. Jackson and H. H. Mantsch), pp. 220-229. SPIE, San Jose, California, USA.
Goodacre, R., Timmins, É. M., Burton, R., Kaderbhai, N., Woodward, A., Kell, D. B. & Rooney, P. J. (1998b). Rapid identification of urinary tract infection bacteria using hyperspectral, whole
organism fingerprinting and artificial neural networks. Microbiol. 144, 1157-1170. [abstract]
Goodacre, R., Timmins, É. M., Rooney, P. J., Rowland, J. J. & Kell, D. B. (1996b). Rapid identification of Streptococcus and Enterococcus species using diffuse reflectance-absorbance Fourier transform infrared spectroscopy and artificial neural networks. FEMS Microbiol. Lett. 140, 233-239. [abstract] [full text]
Goodacre, R., Trew, S., Wrigley-Jones, C., Neal, M. J., Maddock, J., Ottley, T. W., Porter, N. & Kell, D. B. (1994b). Rapid screening for metabolite overproduction in fermentor broths, using pyrolysis mass spectrometry with multivariate calibration and artificial neural networks. Biotechnol. Bioeng. 44, 1205-1216. [full paper]
Heinrich, R. & Rapoport, T. A. (1973). Linear theory of enzymatic chains: its application for the analysis of the crossover theorem and of the glycolysis of human erythrocytes. Acta Biol. Med. Germ. 31, 479-94.
Heinrich, R. & Rapoport, T. A. (1974). A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 42, 89-95.
Heinrich, R. & Schuster, S. (1996). The Regulation of Cellular Systems. Chapman & Hall, New York. [synopsis]
Herweijer, M. A., Berden, J. A. & Slater, E. C. (1986). Uncoupler-inhibitor titrations of ATP-driven reverse electron-transfer in bovine submitochondrial particles provide evidence for direct interaction between ATPase and NADH-Q oxidoreductase. Biochim. Biophys. Acta 849, 276-287. [abstract]
Hieter, P. & Boguski, N. (1997). Functional genomics: it's all how you read it. Science 278, 601-602.
Hinton, J. C. D. (1997). The Escherichia coli genome sequence: the end of an era or the start of the FUN? Mol. Microbiol. 26, 417-422.
Hitchens, G. D. & Kell, D. B. (1983a). On the functional unit of energy coupling in photophosphorylation by bacterial chromatophores. Biochim. Biophys. Acta 723, 308-316.
Hitchens, G. D. & Kell, D. B. (1983b). Uncouplers can shuttle rapidly between localised energy coupling sites during photophosphorylation by chromatophores of Rhodopseudomonas capsulata N22. Biochem. J. 212, 25-30. [abstract]
Ho, M. W. (1998). The Rainbow and the Worm: the Physics of Organisms, Second edition. World Scientific, Singapore. [synopsis]
Hofmeyr, J.-H. S., Kacser, H. & van der Merwe, K. J. (1986). Metabolic control analysis of moiety-conserved cycles. Eur. J. Biochem. 155, 631-641. [abstract]
Holms, H. (1996). Flux analysis and control of the central metabolic pathways in Escherichia coli. FEMS Microbiol. Rev. 19, 85-116. [abstract]
Holms, W. H., Hamilton, I. D. & Mousdale, D. (1991). Improvements to microbial productivity by analysis of metabolic fluxes. J. Chem. Technol. Biotechnol. 50, 139-141.
Kacser, H. (1983). The control of enzyme systems in vivo: elasticity analysis of the steady state. Biochem. Soc. Trans. 11, 35-40.
Kacser, H. & Acerenza, L. (1993). A universal method for achieving increases in metabolite production. Eur. J. Biochem. 216, 361-367. [abstract]
Kacser, H. & Burns, J. A. (1973). The control of flux. In Rate Control of Biological Processes. Symposium of the Society for Experimental Biology Vol 27 (ed. D. D. Davies), pp. 65-104. Cambridge
University Press, Cambridge.
Kaprelyants, A. S. & Kell, D. B. (1992). Rapid assessment of bacterial viability and vitality using rhodamine 123 and flow cytometry. J. Appl. Bacteriol. 72, 410-422.
Kaprelyants, A. S. & Kell, D. B. (1993). Dormancy in stationary-phase cultures of Micrococcus luteus: flow cytometric analysis of starvation and resuscitation. Appl. Environ. Microbiol. 59, 3187-3196.
Kaprelyants, A. S., Mukamolova, G. V. & Kell, D. B. (1994). Estimation of dormant Micrococcus luteus cells by penicillin lysis and by resuscitation in cell-free spent medium at high dilution. FEMS Microbiol. Lett. 115, 347-352.
Kaprelyants, A. S., Mukamolova, G. V., Kormer, S. S., Weichart, D. H., Young, M. & Kell, D. B. (1999). Intercellular signalling and the multiplication of prokaryotes: bacterial cytokines. Symp. Soc. Gen. Microbiol. 57, 33-69. [meeting announcement]
Kell, D. B. (1979). On the functional proton current pathway of electron transport phosphorylation: an electrodic view. Biochim. Biophys. Acta 549, 55-99.
Kell, D. B. (1987). Forces, fluxes and the control of microbial growth and metabolism. The twelfth Fleming lecture. J. Gen. Microbiol. 133, 1651-1665.
Kell, D. B. (1988). Protonmotive energy-transducing systems: some physical principles and experimental approaches. In Bacterial Energy Transduction (ed. C. J. Anthony), pp. 429-490. Academic Press, London.
Kell, D. B. (1992). The protonmotive force as an intermediate in electron transport-linked phosphorylation: problems and prospects. Curr. Top. Cell. Reg. 33, 279-289.
Kell, D. B. (1998). Fom code to mode for orphan genes. Trends Biotechnol. 16, 491-493.
Kell, D. B. & Hitchens, G. D. (1983). Coherent properties of the membranous systems of electron transport phosphorylation. In Coherent Excitations in Biological Systems (ed. H. Fröhlich and F.
Kremer), pp. 178-198. Springer-Verlag, Berlin.
Kell, D. B., Kaprelyants, A. S. & Grafen, A. (1995). On pheromones, social behaviour and the functions of secondary metabolism in bacteria. Trends Ecol. Evol. 10, 126-129.
Kell, D. B., Kaprelyants, A. S., Weichart, D. H., Harwood, C. L. & Barer, M. R. (1998). Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie van Leeuwenhoek 73, 169-187. [abstract]
Kell, D. B., Ryder, H. M., Kaprelyants, A. S. & Westerhoff, H. V. (1991). Quantifying heterogeneity: Flow cytometry of bacterial cultures. Antonie van Leeuwenhoek 60, 145-158. [
abstract]
Kell, D. B. & Sonnleitner, B. (1995). GMP - Good Modelling Practice: an essential component of good manufacturing practice. Trends Biotechnol. 13, 481-492.
Kell, D. B., van Dam, K. & Westerhoff, H. V. (1989). Control analysis of microbial growth and productivity. Symp. Soc. Gen. Microbiol. 44, 61-93.
Kell, D. B. & Welch, G. R. (1991). No turning back, Reductonism and Biological Complexity. Times Higher Educational Supplement 9th August, 15.
Kell, D. B. & Westerhoff, H. V. (1986a). Metabolic control theory: its role in microbiology and biotechnology. FEMS Microbiol. Rev. 39, 305-320.
Kell, D. B. & Westerhoff, H. V. (1986b). Towards a rational approach to the optimization of flux in microbial biotransformations. Trends Biotechnol. 4, 137-142.
Kell, D. B. & Westerhoff, H. V. (1990). Control analysis of organised multienzyme systems. In Structural and organizational aspects of metabolic regulation (ed. P. Srere, M. E. Jones and C. Mathews),
pp. 273-289. Alan R. Liss, New York.
Kholodenko, B. N., Cascante, M., Hoek, J. B., Westerhoff, H. V. & Schwaber, J. (1998). Metabolic design: How to engineer a living cell to desired metabolite concentrations and fluxes. Biotechnol.
Bioeng. 59, 239-247.
Koonin, E. V. & Galperin, M. Y. (1997a). Prokaryotic genomes: the emerging paradigm of genome-based microbiology. Curr. Op. Gen. & Develop. 7, 757-763. [abstract]
Koonin, E. V., Mushegian, A. R., Galperin, M. Y. & Walker, D. R. (1997b). Comparison of archaeal and bacterial genomes: computer analysis of protein sequences predicts novel functions and suggests a chimeric origin for the archaea. Mol. Microbiol. 25, 619-637. [abstract]
Langley, P., Simon, H. A., Bradshaw, G. L. & Zytkow, J. M. (1987). Scientific Discovery: Computational exploration of the creative processes. MIT Press, Cambridge, MA. [MIT Press description]
Letellier, T., Malgat, M., Rossignol, R. & Mazat, J. P. (1998). Metabolic control analysis and mitochondrial pathologies. Mol. Cell. Biochem. 184, 409-417. [abstract]
Lopez de Felipe, F., Kleerebezem, M., de Vos, W. M. & Hugenholtz, J. (1998). Cofactor engineering: a novel approach to metabolic engineering in Lactococcus lactis by controlled expression of NADH oxidase. J. Bacteriol. 180, 3804-3808. [abstract]
Mazat, J. P., Letellier, T., Bedes, F., Malgat, M., Korzeniewski, B., Jouaville, L. S. & Morkuniene, R. (1997). Metabolic control analysis and threshold effect in oxidative phosphorylation: Implications for mitochondrial pathologies. Mol. Cell. Biochem. 174, 143-148. [abstract]
Mazat, J. P., Letellier, T., Malgat, M., Rossignol, R., Korzeniewski, B., Demaugre, F. & Leroux, J. P. (1998). Inborn errors of metabolism in the light of metabolic control analysis. Biochem. Soc. Trans. 26, 141-145.
McVeigh, H. P., Munro, J. & Embley, T. M. (1996). Molecular evidence for the presence of novel actinomycete lineages in a temperate forest soil. J. Ind. Microbiol. 17, 197-204. [abstract]
Mendes, P. (1993). GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput. Appl. Biosci. 9, 563-571. [abstract]
Mendes, P. (1997). Biochemistry by numbers: simulation of biochemical pathways with Gepasi 3. Trends Biochem. Sci. 22, 361-363.
Mendes, P. & Kell, D. B. (1997). Making cells work: metabolic engineering for everyone. Trends Biotechnol. 15, 6-7.
Mendes, P. & Kell, D. B. (1998a). Non-linear optimization of biochemical pathways: applications to metabolic engineering and parameter estimation. Bioinformatics 14, 869-883. [abstract]
Mendes, P. & Kell, D. B. (1998b). Numerical optimisation and simulation for rational metabolic engineering. In Biothermokinetics in the post genomic era (ed. C. Larsson, I.-L. Pahlman and L. Gustaffson), pp. 345-349. Chalmers Repro Service, Göteborg.
Mendes, P., Kell, D. B. & Welch, G. R. (1995). Metabolic channeling in organized enzyme systems: experiments and models. In Enzymology in vivo (ed. K. M. Brindle), pp. 1-19. JAI Press, London.
Mendes, P., Kell, D. B. & Westerhoff, H. V. (1992). Channelling can decrease pool size. Eur. J. Biochem. 204, 257-266. [abstract]
Mendes, P., Kell, D. B. & Westerhoff, H. V. (1996). Why and when channeling can decrease pool size at constant net flux in a simple dynamic channel. Biochim. Biophys. Acta 1289, 175-186. [abstract]
Michie, D., Spiegelhalter, D. J. & Taylor, C. C. (1994). Machine learning: neural and statistical classification. In Ellis Horwood Series in Artificial Inteligence (ed. J. Campbell). Ellis Horwood,
Chichester. [full text]
Mitchell, P. (1966). Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. 41, 445-502.
Mukamolova, G. V., Kaprelyants, A. S., Young, D. I., Young, M. & Kell, D. B. (1998). A bacterial cytokine. Proc. Natl. Acad. Sci. USA 95, 8916-8921. [abstract]
Nicholls, D. G. & Ferguson, S. J. (1992). Bioenergetics 2. Academic Press, London. [
synopsis]
Niederberger, P., Prasad, R., Miozzari, G. & Kacser, H. (1992). A Strategy for increasing an in vivo flux by genetic manipulations - the tryptophan system of yeast. Biochem. J. 287, 473-479. [abstract]
Oldroyd, D. (1986). The arch of knowledge: an introduction to the history of the philosophy and methodology of science. Methuen, New York.
Oliver, S. G. (1996). From DNA sequence to biological function. Nature 379, 597-600. [abstract]
Oliver, S. G. & Baganz, F. (1998). The yeast genome: systematic analysis of DNA sequence and biological function. In Genomics: commercial opportunities from a scientific revolution (ed. L. G. Copping, G. K. Dixon and D. J. Livingstone), pp. in press. Bios, Oxford.
Oliver, S. G., Winson, M. K., Kell, D. B. & Baganz, F. (1998). Systematic functional analysis of the yeast genome. Trends Biotechnol. 16, 373-378. [abstract]
Ovádi, J. (1995). Cell architecture and metabolic channeling. Springer-Verlag, New York. [ entry in Amazon.com]
Raju, G. K. & Cooney, C. L. (1998). Active learning from process data. AIChE J. 44, 2199-2211.
Reder, C. (1988). Metabolic control theory: a structural approach. J. Theoret. Biol. 135, 175-201. [abstract]
Reich, J. G. & Sel'kov, E. E. (1981). Energy metabolism of the cell: a theoretical treatise. Academic Press, London.
Rich, E. & Knight, K. (1991). Artificial Intelligence. McGraw Hill, New York. [synopsis]
Rivera, M. C., Jain, R., Moore, J. E. & Lake, J. A. (1998). Genomic evidence for two functionally distinct gene classes. Proc. Natl. Acad. Sci. USA 95, 6239-6244. [abstract]
Schena, M., Shalon, D., Heller, R., Chai, A., Brown, P. O. & Davis, R. W. (1996). Parallel human genome analysis - microarray-based expression monitoring of 1000 genes. Proc. Natl. Acad. Sci. USA 93, 10614-10619. [abstract]
Schlosser, P. M., Holcomb, T. & Bailey, J. E. (1993). Determining metabolic sensitivity coefficients directly from experimental data. Biotechnol. Bioeng. 41, 1027-1038.
Schuster, R. & Holzhütter, H. G. (1995). Use of mathematical models for predicting the metabolic effect of large-scale enzyme-activity alterations: application to enzyme deficiencies of red blood cells. Eur. J. Biochem. 229, 403-418. [abstract]
Shaw, A. D., diCamillo, A., Vlahov, G., Jones, A., Bianchi, G., Rowland, J. & Kell, D. B. (1997). Discrimination of the variety and region of origin of extra virgin olive oils using C-13 NMR and multivariate calibration with variable reduction. Anal. Chim. Acta 348, 357-374.
Simpson, T. W., Colon, G. E. & Stephanopoulos, G. (1995). 2 Paradigms of metabolic engineering applied to amino acid biosynthesis. Biochem. Soc. Trans. 23, 381-387.
Simpson, T. W., Shimizu, H. & Stephanopoulos, G. (1998). Experimental determination of group flux control coefficients in metabolic networks. Biotechnol. Bioeng. 58, 149-153.
Small, J. R. (1993). Flux control coefficients determined by inhibitor titration: the design and analysis of experiments to minimize errors. Biochem. J. 296, 423-433. [abstract]
Small, J. R. & Kacser, H. (1993a). Responses of metabolic systems to large changes in enzyme activities and effectors .1. The linear treatment of unbranched chains. Eur. J. Biochem. 213, 613-624. [abstract]
Small, J. R. & Kacser, H. (1993b). Responses of metabolic systems to large changes in enzyme activities and effectors .2. The linear treatment of branched pathways and metabolite concentrations - assessment of the general nonlinear case. Eur. J. Biochem. 213, 625-640. [abstract]
Smith, V., Botstein, D. & Brown, P. O. (1995). Genetic footprinting - A genomic strategy for determining a gene's function given its sequence. Proc. Natl. Acad. Sci. USA 92, 6479-6483. [abstract]
Smith, V., Chou, K. N., Lashkari, D., Botstein, D. & Brown, P. O. (1996). Functional analysis of the genes of yeast chromosome V by genetic footprinting. Science 274, 2069-2074. [abstract]
Spellman, P. T., Sherlock, G., Zhang, M. Q., Iyer, V. R., Anders, K., Eisen, M. B., Brown, P. O., Botstein, D. & Futcher, B. (1998). Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9, 3273-3297. [abstract]
Stephanopoulos, G. & Simpson, T. W. (1997). Flux amplification in complex metabolic networks. Chem. Eng. Sci. 52, 2607-2627.
Taylor, J., Goodacre, R., Wade, W. G., Rowland, J. J. & Kell, D. B. (1998). The deconvolution of pyrolysis mass spectra using genetic programming: application to the identification of some Eubacterium species. FEMS Microbiol. Lett. 160, 237-246. [abstract]
Teusink, B., Baganz, F., Westerhoff, H. V. & Oliver, S. G. (1998). Metabolic Control Analysis as a tool in the elucidation of the function of novel genes. In Methods in Microbiology: Yeast gene analysis (ed. M. F. Tuite and A. J. P. Brown), pp. 297-336. Academic Press, London. [book synopsis]
Thatcher, J. W., Shaw, J. M. & Dickinson, W. J. (1998). Marginal fitness contributions of nonessential genes in yeast. Proc. Natl. Acad. Sci. USA 95, 253-257. [abstract]
Thomas, S. & Fell, D. A. (1995). Error and bias in control coefficients calculated from elasticities. Biochem. Soc. Trans. 23, S294.
Thomas, S. & Fell, D. A. (1998). The role of multiple enzyme activation in metabolic flux control. Adv. Enz. Reg. 38, 65-85.
Velculescu, V. E., Zhang, L., Zhou, W., Vogelstein, J., Basrai, M. A., Bassett, D. E., Hieter, P., Vogelstein, B. & Kinzler, K. W. (1997). Characterization of the yeast transcriptome. Cell
88, 243-251. [abstract]
Weiss, S. H. & Kulikowski , C. A. (1991). Computer systems that Learn: classification and prediction methods from statistics, neural networks, machine learning, and expert systems. Morgan Kaufmann Publishers, San Mateo, CA.
Welch, G. R. & Kell, D. B. (1986). Not just catalysts; the bioenergetics of molecular machines. In The Fluctuating Enzyme (ed. G. Welch, .R.), pp. 451-492. John Wiley, New York.
Westerhoff, H. V., Hellingwerf, K. J. & van Dam, K. (1983). Thermodynamic efficiency of microbial growth is low but optimal for maximal growth rate. Proc. Natl. Acad. Sci. USA 80, 305-309.
Westerhoff, H. V. & Kell, D. B. (1987). Matrix method for determining the steps most rate-limiting to metabolic fluxes in biotechnological processes. Biotechnol. Bioeng. 30, 101-107.
Westerhoff, H. V. & Kell, D. B. (1988). A control theoretical analysis of inhibitor titrations of metabolic channelling. Comments Mol. Cell. Biophys. 5, 57-107.
Westerhoff, H. V. & Kell, D. B. (1996). What BioTechnologists knew all along...? J. Theoret. Biol. 182, 411-420. [abstract]
Westerhoff, H. V. & van Dam, K. (1987). Thermodynamics and control of biological free energy transduction. Elsevier, Amsterdam.
Wilkins, M. R., Williams, K. L., Appel, R. D. & Hochstrasser, D. F. (1997). Proteome research: new frontiers in functional genomics. Springer, Berlin. [synopsis]
Williams, R. J. (1956). Biochemical Individuality. John Wiley, New York.
Winson, M. K., Goodacre, R., Timmins, É. M., Jones, A., Alsberg, B. K., Woodward, A. M., Rowland, J. J. & Kell, D. B. (1997). Diffuse reflectance absorbance spectroscopy taking in chemometrics (DRASTIC). A hyperspectral FT-IR-based approach to rapid screening for metabolite overproduction. Anal. Chim. Acta 348, 273-282. [abstract]
Winson, M. K., Todd, M., Rudd, B. A. M., Jones, A., Alsberg, B. K., Woodward, A. M., Goodacre, R., Rowland, J. J. & Kell, D. B. (1998). A DRASTIC (Diffuse Reflectance Absorbance Spectroscopy Taking in Chemometrics) approach for the rapid analysis of microbial fermentation products: quantification of aristeromycin and neplanocin A in Streptomyces citricolor broths. In New frontiers in screening for microbial biocatalysts (ed. K. Kieslich, C. P. van der Beek, J. A. M. de Bont and W. J. J. van den Tweel), pp. 185-191. Elsevier, Amsterdam.
Wodicka, L., Dong, H. L., Mittmann, M., Ho, M. H. & Lockhart, D. J. (1997). Genome-wide expression monitoring in Saccharomyces cerevisiae. Nature Biotechnol. 15, 1359-1367. [abstract]
|
|
Copyright © 1999 Douglas B. Kell and Pedro Mendes, All rights reserved.
Last update: 19 February 2004
|
|
 |
Group |
|
|
|
|
|
Back to the group's homepage.
| |
|
